克利夫兰诊所和 IBM 的研究人员最近在《化学理论与计算杂志》上发表了研究成果,为将量子计算方法应用于蛋白质结构预测奠定了基础。
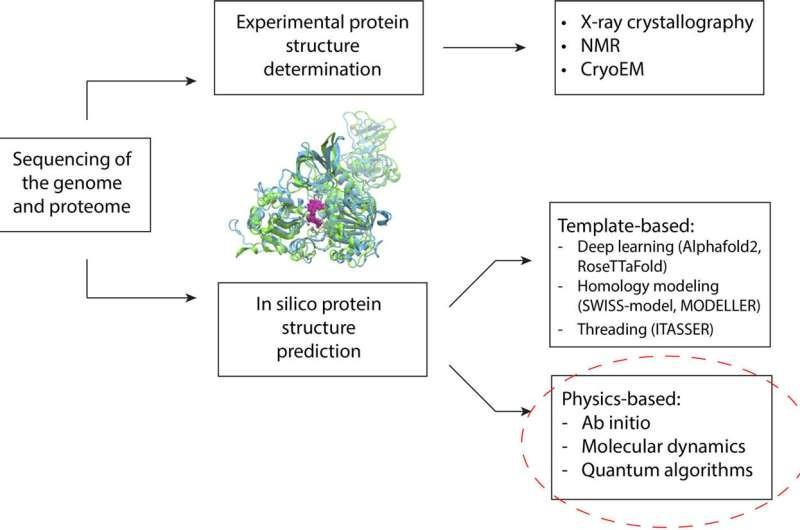
几十年来,研究人员一直利用计算方法来预测蛋白质结构。蛋白质折叠成一种结构,决定了它如何发挥作用以及如何与体内其他分子结合。这些结构决定了人类健康和疾病的许多方面。
通过准确预测蛋白质的结构,研究人员可以更好地了解疾病如何传播,从而开发有效的治疗方法。克利夫兰诊所博士后研究员 Bryan Raubenolt 博士和 IBM 研究员 Hakan Doga 博士带领团队探索量子计算如何改进当前方法。
近年来,机器学习技术在蛋白质结构预测方面取得了重大进展。这些方法依赖于训练数据(通过实验确定的蛋白质结构的数据库)来进行预测。这意味着它们受到它们被教导识别的蛋白质数量的限制。当程序/算法遇到突变的蛋白质或与它们所训练的蛋白质有很大不同(这在遗传疾病中很常见)的蛋白质时,这会导致准确度降低。
另一种方法是模拟蛋白质折叠的物理原理。通过模拟,研究人员可以观察给定蛋白质的各种可能形状,并找到最稳定的形状。最稳定的形状对于药物设计至关重要。
挑战在于,这些模拟在传统计算机上几乎不可能进行,只要蛋白质尺寸超过一定值。在某种程度上,增加目标蛋白质的尺寸就相当于增加魔方的尺寸。对于含有 100 个氨基酸的小蛋白质,传统计算机需要相当于宇宙年龄的时间来彻底搜索所有可能的结果,Raubenolt 博士说。
为了克服这些限制,研究团队采用了量子和经典计算方法的混合方法。该框架可以让量子算法解决对最先进的经典计算具有挑战性的领域,包括蛋白质大小、内在无序性、突变和蛋白质折叠所涉及的物理学。与最先进的经典方法相比,该框架通过量子计算机上准确预测寨卡病毒蛋白质小片段的折叠得到了验证。
量子-经典混合框架的初步结果优于基于经典物理的方法和 AlphaFold2。尽管后者的设计最适合处理较大的蛋白质,但它仍然证明了该框架能够在不直接依赖大量训练数据的情况下创建精确模型的能力。
研究人员首先使用量子算法对片段主链的最低能量构象进行建模,这通常是计算中最耗费计算资源的步骤。然后使用经典方法转换从量子计算机获得的结果,重建带有侧链的蛋白质,并使用经典分子力学力场对结构进行最终细化。
该项目展示了将问题分解成各个部分的方法之一,利用量子计算方法解决其中的一些部分,利用经典计算解决其他部分,以提高准确性。
“该项目最独特的一点是涉及的学科数量,”Raubenolt 博士说。“我们团队的专业知识范围广泛,从计算生物学和化学、结构生物学、软件和自动化工程到实验原子和核物理学、数学,当然还有量子计算和算法设计。我们利用这些领域的知识来创建一个计算框架,可以模拟人类生命中最重要的过程之一。”
该团队将经典计算方法与量子计算方法相结合,是加深我们对蛋白质结构及其如何影响我们治疗和预防疾病能力的理解的重要一步。该团队计划继续开发和优化量子算法,以预测更大、更复杂的蛋白质结构。
Doga 博士表示:“这项研究是探索量子计算能力在蛋白质结构预测中的优势的重要一步。我们的目标是设计出能够尽可能真实地预测蛋白质结构的量子算法。”
本站全部资讯来源于实验室原创、合作机构投稿及网友汇集投稿,仅代表个人观点,不作为任何依据,转载联系作者并注明出处:https://www.lvsky.net/281.html
评论