蛋白质是所有生物体中必不可少的分子。它们在我们身体的结构和功能中发挥着核心作用,也出现在我们每天接触的许多产品中,从药物到洗衣粉等家居用品。每种蛋白质都是氨基酸构建块的链条,就像图像可能包含多个物体(如狗和猫)一样,蛋白质也可能有多个组成部分,这些组成部分称为蛋白质结构域。了解蛋白质的氨基酸序列(例如其结构域)与其结构或功能之间的关系是一项长期存在的挑战,具有深远的科学意义。
一个已知结构的蛋白质示例,来自大肠杆菌的 TrpCF,其用于预测功能的区域以绿色突出显示。这种蛋白质产生色氨酸,而色氨酸是人体饮食中必不可少的一部分。
很多人都熟悉通过氨基酸序列计算预测蛋白质结构的最新进展,例如 DeepMind 的AlphaFold。同样,科学界也有着长期使用计算工具直接从序列推断蛋白质功能的历史。例如,广泛使用的蛋白质家族数据库Pfam包含大量高度详细的计算注释,用于描述蛋白质结构域的功能,如珠蛋白和胰蛋白酶家族。虽然现有方法已经成功预测了数亿种蛋白质的功能,但仍有许多蛋白质的功能未知 — — 例如,至少三分之一的微生物蛋白质没有可靠的注释。随着公共数据库中蛋白质序列的数量和多样性持续快速增加,准确预测高度发散序列的功能的挑战变得越来越紧迫。
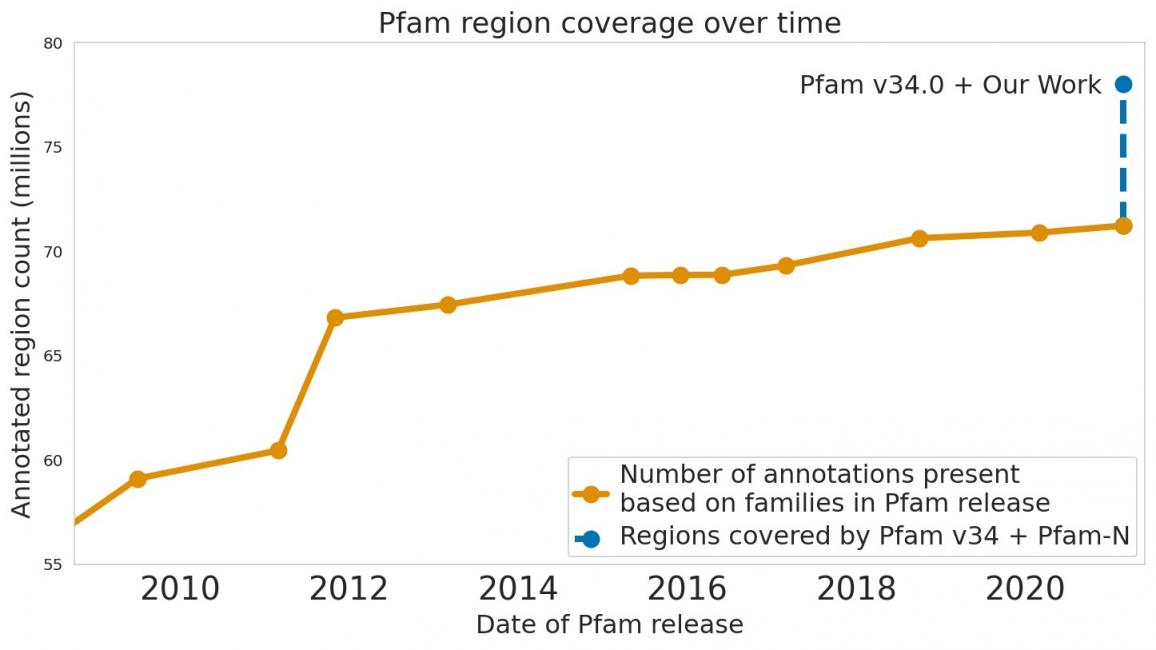
在《自然生物技术》杂志上发表的 《利用深度学习注释蛋白质世界》中,我们描述了一种可靠地预测蛋白质功能的机器学习 (ML) 技术。这种方法被我们称为 ProtENN,它使我们能够向 Pfam 著名且值得信赖的蛋白质功能注释集添加约 680 万个条目,大约相当于过去十年的进展总和,我们将其作为Pfam-N发布。为了鼓励朝这个方向进一步研究,我们发布了 ProtENN 模型和一篇类似extract的交互式文章,研究人员可以在其中试验我们的技术。这个交互式工具允许用户在浏览器中输入序列并实时获得预测蛋白质功能的结果,无需任何设置。在这篇文章中,我们将概述这一成就以及我们在揭示更多蛋白质世界方面取得的进展。
Pfam 数据库包含大量蛋白质家族及其序列。我们的 ML 模型 ProtENN 帮助注释了数据库中 680 多万个蛋白质区域。
蛋白质功能预测作为分类问题
在计算机视觉领域,通常先训练一个用于图像分类任务的模型,例如CIFAR-100,然后再将其扩展到更专业的任务,例如对象检测和定位。同样,我们开发了一个蛋白质域分类模型,作为未来用于整个蛋白质序列分类的模型的第一步。我们将问题定义为一个多类别分类任务,其中给定蛋白质域的氨基酸序列,我们从 17,929 个类别(Pfam 数据库中包含的所有类别)中预测单个标签。
将序列与功能联系起来的模型
虽然目前有许多可用于蛋白质域分类的模型,但目前最先进的方法的一个缺点是它们基于线性序列的比对,不考虑蛋白质序列不同部分氨基酸之间的相互作用。但蛋白质不仅仅是一排氨基酸,它们会自行折叠,因此不相邻的氨基酸会相互产生强烈影响。
将新的查询序列与一个或多个已知功能的序列比对是当前最先进方法的关键步骤。这种对已知功能序列的依赖使得如果新序列与任何已知功能的序列高度不同,则很难预测其功能。此外,基于比对的方法计算量大,并且将其应用于大型数据集(例如包含超过 10 亿个蛋白质序列的宏基因组数据库MGnify)的成本可能过高。
为了应对这些挑战,我们建议使用扩张 卷积神经网络(CNN),这种神经网络非常适合对非局部成对氨基酸相互作用进行建模,并且可以在 GPU 等现代 ML 硬件上运行。我们训练一维 CNN 来预测蛋白质序列的分类,我们将其称为 ProtCNN,以及一组独立训练的 ProtCNN 模型,我们将其称为 ProtENN。我们使用这种方法的目标是通过开发一种可靠的 ML 方法来补充传统的基于比对的方法,从而为科学文献增加知识。为了证明这一点,我们开发了一种方法来准确测量我们方法的准确性。
以进化为出发点进行评估
与其他领域中众所周知的分类问题类似,蛋白质功能预测的挑战不在于开发一个全新的模型,而在于创建公平的训练和测试集,以确保模型能够对未知数据做出准确的预测。由于蛋白质是从共同的祖先进化而来的,不同的蛋白质通常共享相当一部分氨基酸序列。如果不加以适当的处理,测试集可能会被与训练数据高度相似的样本所主导,这可能导致模型仅仅通过“记住”训练数据就能表现良好,而不是学会从中更广泛地概括。
我们创建了一个测试集,要求 ProtENN 能够很好地概括远离其训练集的数据。
为了防止这种情况发生,必须使用多个单独的设置来评估模型性能。对于每次评估,我们根据每个保留的测试序列与训练集中最近的序列之间的相似性对模型准确率进行分层。
第一次评估包括聚类分割训练集和测试集,与先前文献一致。在这里,蛋白质序列样本按序列相似性聚类,整个簇被放入训练集或测试集中。因此,每个测试示例与每个训练示例至少有 75% 的不同。这项任务的出色表现表明模型可以推广,以对分布外的数据做出准确的预测。
对于第二次评估,我们使用随机分割的训练和测试集,根据对样本分类难度的估计对样本进行分层。这些难度指标包括:(1) 测试样本与最接近的训练样本之间的相似性,以及 (2) 来自真实类别的训练样本数量(仅使用少量训练样本来准确预测函数要困难得多)。
为了将我们的工作放在背景中,我们评估了最广泛使用的基线模型和评估设置的性能,特别是以下基线模型:(1)BLAST,一种使用序列比对来测量距离和推断功能的最近邻方法,以及(2)概要隐马尔可夫模型(TPHMM 和 phmmer)。对于其中每一个,我们都包括基于上述序列比对相似性的模型性能分层。我们将这些基线与 ProtCNN 和 CNN 集合 ProtENN 进行了比较。
我们衡量每个模型的概括能力,从最难的例子(左)到最简单的例子(右)。
可重复且可解释的结果
我们还与 Pfam 团队合作,他们是来自欧洲分子生物学实验室欧洲生物信息学研究所(EMBL-EBI) 的国际知名专家,以测试我们的方法论概念验证是否可用于标记真实世界序列。我们证明了ProtENN 可以学习与基于比对的方法互补的信息,并创建了两种方法的组合来标记比任何一种方法单独标记的序列更多的序列。我们公开发布了这项工作的结果Pfam-N,这是一组 680 万个新的蛋白质序列注释。
在看到这些方法和分类任务的成功后,我们检查了这些网络,以了解嵌入是否普遍有用。我们构建了一个工具,使用户能够探索模型预测、嵌入和输入序列之间的关系,我们已通过交互式手稿提供该工具,我们发现相似的序列在嵌入空间中聚集在一起。此外,我们选择的网络架构(扩张的 CNN)使我们能够采用先前发现的可解释性方法,例如类激活映射(CAM) 和足够的输入子集(SIS),以识别负责神经网络预测的子序列。通过这种方法,我们发现我们的网络通常关注序列的相关元素来预测其功能。
结论和未来工作
我们很高兴看到过去几年来,通过将机器学习应用于理解蛋白质结构和功能所取得的进展,这反映在更广泛的研究界的贡献中,从AlphaFold和CAFA到会议上专门讨论这一主题的众多研讨会和研究报告。在我们期待进一步开展这项工作时,我们认为继续与该领域的科学家合作,分享他们的专业知识和数据,再加上机器学习的进步,将有助于我们进一步揭示蛋白质世界。
致谢
我们要感谢手稿的所有合著者,Maysam Moussalem、Jamie Smith、Eli Bixby、Babak Alipanahi、Shanqing Cai、Cory McLean、Abhinay Ramparasad、Steven Kearnes、Zack Nado 和 Tom Small。 此外,我们还要感谢 EMBL-EBI 的 Pfam 团队在发布 Pfam-N 方面的合作。
评论